cfSNV is an ultra-sensitive and accurate somatic SNV caller designed for cfDNA sequencing. Taking advantage of modern statistical models and machine learning approaches, cfSNV provides hierarchical mutation profiling and multi-layer error suppression, including error suppression in read mates, site-level error filtration and read-level error filtration. The R package of cfSNV is available on GitHub. In addition, we also implemented a Docker image of the cfSNV package, which is designed such that researchers and clinicians with a limited computational background can easily carry out analyses on both high-performance computing platforms and local computers. The docker image is available on GitHub as well. Both R package and Docker image of cfSNV can be freely used for educational and research purposes by non-profit institutions and U.S. government agencies only under the UCLA Academic Software License.
CancerDetector is a software package for ultrasensitively detecting tiny amount of tumor-derived cell-free DNA out of total cell-free DNAs in plasma, using the DNA methylation profiles of cell-free DNAs. The key of this method is to probabilistically model the joint methylation patterns of multiple adjacent CpG sites on an individual sequencing read, in order to exploit the pervasive nature of DNA methylation for signal amplification. Therefore, CancerDetector can sensitively identify a trace amount of tumor cfDNAs in plasma, at the level of individual reads. The software package and source codes of CancerDetector are available here.
CancerLocator is a software package for non-invasive cancer diagnosis using methylation profiles of cell-Free DNA. It exploits the diagnostic potential of cell-free DNA by determining not only the presence but also the location of tumors. CancerLocator simultaneously infers the proportions and the tissue-of-origin of tumor-derived cell-free DNA in a blood sample using genome-wide DNA methylation data.CancerLocator is implemented in Java and is freely available on GitHub GitHub under the MIT license. The source code is also available at Zenodo (DOI: 10.5281/zenodo.375649).
Three-dimensional (3D) genome structures vary from cell to cell even in an isogenic sample. Unlike protein structures, genome structures are highly plastic, posing a significant challenge for structure-function mapping. Struct2fun is a software tool to comprehensively identify 3D chromatin clusters that each occurs frequently across a population of genome structures, either deconvoluted from ensemble-averaged Hi-C data or from a collection of single-cell Hi-C data. The software package and source codes of Struct2fun are available here.
TopDom is an efficient and deterministic tool to identify topological domains, along with a set of statistical methods for evaluating their quality. TopDom is much more efficient than existing methods and depends on just one intuitive parameter, a window size, for which we provide easy-to-implement optimization guidelines. The software package and source codes of TopDom are available Zhoulab Github.
Hi-Corrector is a fast, scalable and memory-efficient software package to remove biases from chromatin contact matrices generated by genome-wide proximity ligation assays, e.g. Hi-C and its variant TCC. The sequential version is implemented in ANSI C and can be easily compiled on any system; the parallel version is implemented in ANSI C with the MPI library (a standardized and portable parallel environment designed for solving large-scale scientific problems). The software package and source codes of Hi-Corrector are available Zhoulab Github.
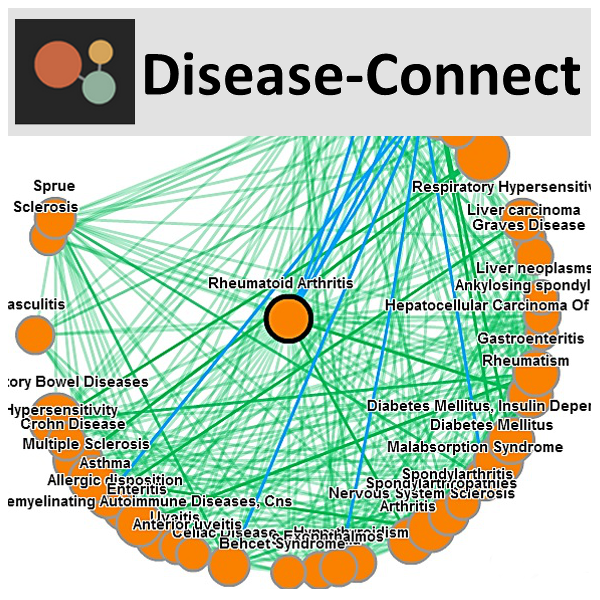
The DiseaseConnect is the first public web server for analysis and visualization of a comprehensive knowledge on mechanism-based disease connectivity, by integrating comprehensive omics and literature data, including a large amount of Genome-Wide Association Studies (GWAS) catalog, gene expression data, and text-mined knowledge, to discover disease-disease connectivity via common molecular mechanisms. The clinical co-morbidity data and a comprehensive compilation of known drug-disease relationships are also supplemented for understanding the disease landscape and for facilitating the mechanism-based development of new drug treatments. [know more]

iArray is a data mining and visualization software platform for the integrative analysis of multiple cross-platform microarray datasets. Due to the noisy nature of microarray data, identifying recurrent signals across several datasets could enhance signal to noise separation, and allow us to draw biological inference with higher confidence. [know more]
CODENSE is a software package to mine coherent dense subgraphs from multiple biological networks. CODENSE is short for Mining Coherent Dense Subgraphs. By simplifying the problem of identifying coherent dense subgraphs across n graphs into a problem of identifying dense subgraphs in two special graphs: the summary graph and the second-order graph, CODENSE can efficiently mine frequent coherent dense subgraphs across large numbers of massive graphs. [know more]
MODES is short for Mining Overlapping DENSE Subgraphs. MODES is developed based on HCS (Mining Highly Connected Subgraphs) (Hartuv & Shamir, 2000), with two new features: (1) MODES is more efficient in identifying dense subgraphs; and more importantly, (2) MODES can discover overlapping subgraphs. MODES is used in CODENSE to identify dense subgraphs. [know more]